


近日,一则“女童肚子鼓如球”的新闻被曝光,在网上引起一片热议。照片里,年仅3岁的孩子不但体重不如同龄人(仅11公斤,脾脏估计就有5公斤),就连身高也比同龄孩子要矮上一大截。
而腹部却鼓得如一个大皮球,肚皮上还布满了血筋,像极了一个足月了快要生产的孕妇,但鼓得历害的肚子,好像是随时都有可能被撑破的风险。
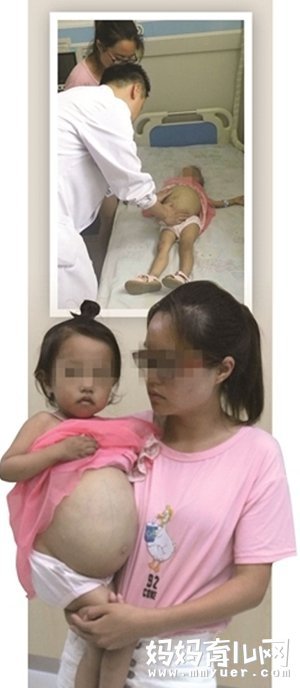
这不看不知道,一看还吓一跳。原来这小女孩得了一种叫做戈谢病的罕见疾病。那么到底这个戈谢病是个什么东西?今天我们就一起来看看。
首先我们要知道,“戈谢病”是罕见病中的罕见病,就我国目前而言,全国确诊的只有300多人。据悉,任何年龄自出生至80岁均可发病,但以少年儿童多发,7岁以下更多。患此病的人内脏肿大,直接表现就是大肚子,会导致疼痛、骨损伤甚至死亡。
戈谢病可分为三型:
成人型(Ⅰ型)
为本病最常见类型,也是脂质贮积病中常见者。犹太人(Ashkenazi-Jewish民族)中多见,但各民族中均有。在美国估计每年儿童患者不到5000例。任何年龄均可起病,常以脾脏大就医。进展可快可慢,进展慢者,脾脏大尤甚,有时有脾梗死或脾破裂而发生急腹症症状。肝脏呈进行性肿大,但不如脾脏肿大明显。病程久者,皮肤及黏膜呈茶黄色,常误诊为黄疸,暴露部位如颈、手及小腿最明显,呈棕黄色。
眼球结膜上常有楔形睑裂斑,底在角膜边缘,尖指向内、外眦,初呈黄白色,后变为棕黄色。肺累及时可影响气体交换而出现症状。晚期患者四肢可有骨痛,甚而病理性骨折,以股骨下端最常见,也可累及股骨颈及脊柱骨。有脾功能亢进时可因血小板减少而有出血倾向。小儿患者身高及体重常受影响。
婴儿型(Ⅱ型)
患儿自生后即可有肝大、脾大,3~6个月时已很明显,有吸吮、吞咽困难,生长发育落后表现。神经系统症状突出,颈强直、头后仰、肌张力增高、角弓反张、踺反射亢进,最后变为软瘫,无反应。脑神经受累时可有内斜,面瘫等症状。易并发感染。由于病程短暂,多于婴儿期死亡,因此肝、脾脏肿大不如成人型明显,无皮肤色素沉着,骨骼改变不显著。
幼年型(Ⅲ型)
常于2岁至青少年期发病,脾大常于体检时发现,一般呈中度肿大。病情进展缓慢,逐渐出现中枢神经系统症状,如:肌阵挛性抽搐、动作不协调、精神错乱,最后卧床不起。肝脏常轻微肿大,但也可进行性肿大而出现肝功能严重损害。
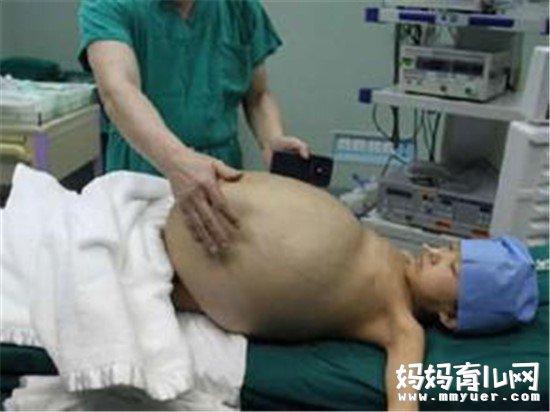
那么如此可怕的疾病到底是什么引起的呢?
据悉戈谢病为常染色体隐性遗传性疾病。一种最常见的溶酶体贮积症,属于常染色体隐性方式遗传,因位于1 号染色体长臂上的葡萄糖脑苷脂酶基因的突变,造成体内该酶的缺乏,细胞内脂类分子无法正常代谢而堆积在溶酶体中。导致各种临床表现如肝脾肿大、贫血、血小板减少以及骨骼病变等各种临床表现。
此病是否有药可医
酶替代疗法是目前国内治疗戈谢病的新标准。具体做法是通过两周一次的常规静脉滴注来替代缺少的酶。
据介绍,注射用的伊米苷酶即“思而赞”,是我国目前唯一能有效治疗戈谢病的药物。但这种药物价格不菲。而且,戈谢病患者的用药剂量需要根据患者的体重以及疾病的严重程度。按照正常剂量,现在熙熙每月需要注射2支药,这样算下来,差不多每年需要60万元的治疗费用,并且随着年龄的增长,孩子用药量要增加,每年的治疗费用或超过百万。
如此昂贵的费用,对于一个家庭来讲是可怕的。但是,据悉,上海、昆明、宁夏、青岛等地,已经出台了针对戈谢病患者的医疗保障政策。2016年1月1日起,浙江也将戈谢病纳入了医保。
医生呼吁,希望政府尽早将戈谢病纳入医保范畴,从制度和政策上保证戈谢病群体的长期治疗。要知道对于戈谢病人来说,只要终身用药,生活就和正常人无异,而这对于戈谢病家庭来说,却是弥足珍贵的。











 正在加载中...
正在加载中... 






